Genome-wide DNA methylation profiling of non-small cell lung carcinomas
Peer reviewed, Journal article
Peer reviewed

Åpne
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Permanent lenke
https://hdl.handle.net/1956/8714Utgivelsesdato
2012-06-22Metadata
Vis full innførselSamlinger
Originalversjon
https://doi.org/10.1186/1756-8935-5-9Sammendrag
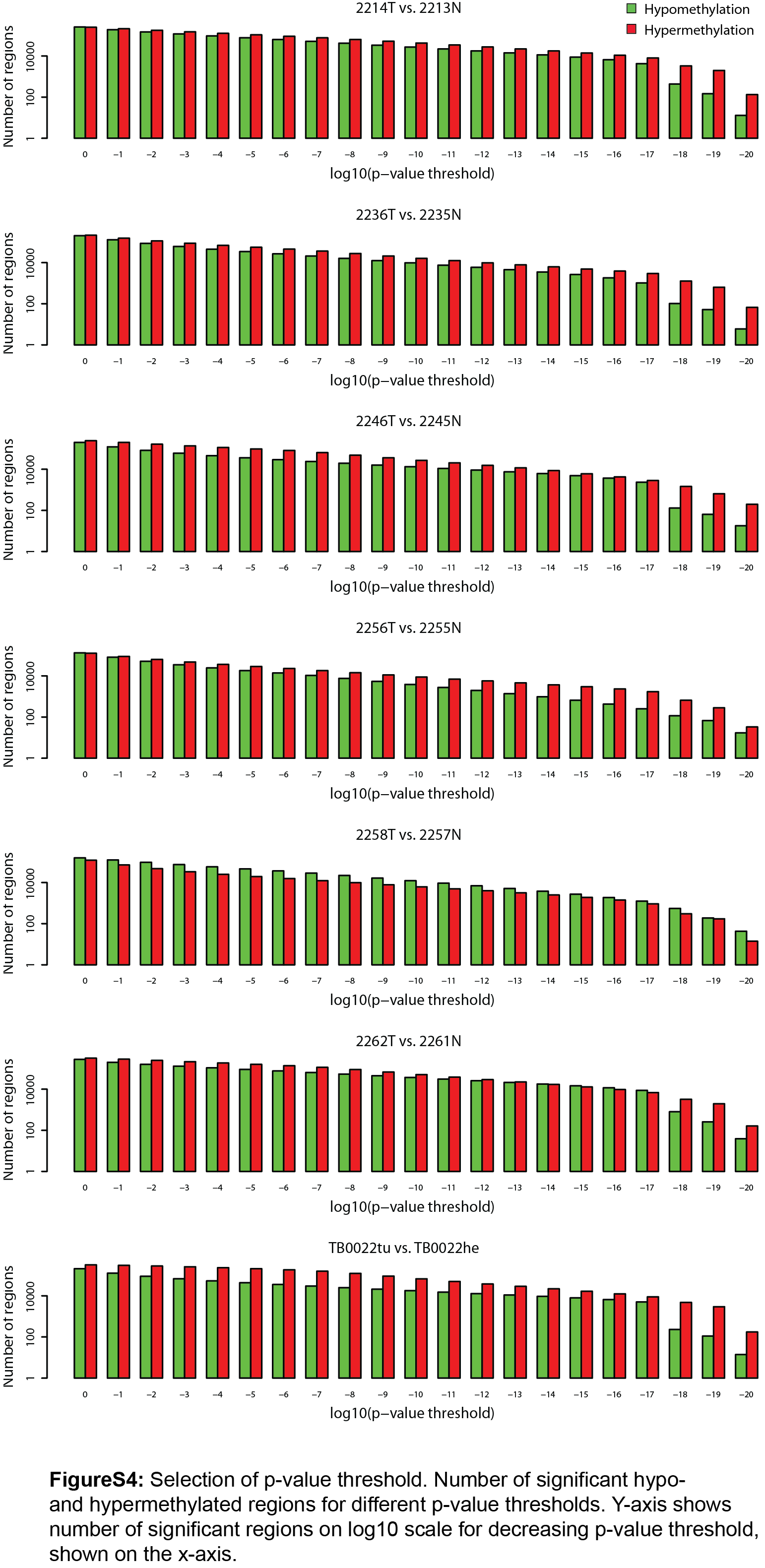
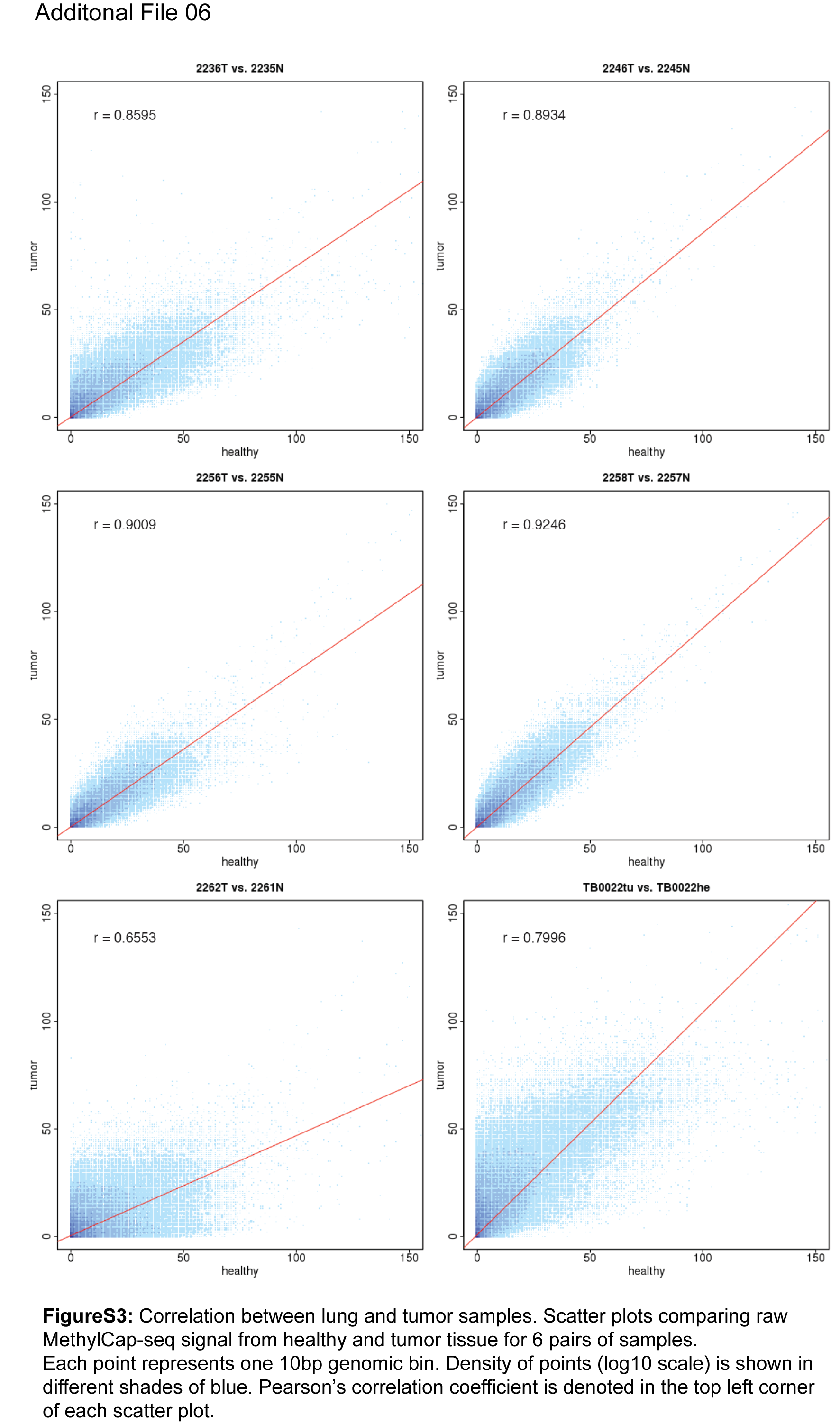
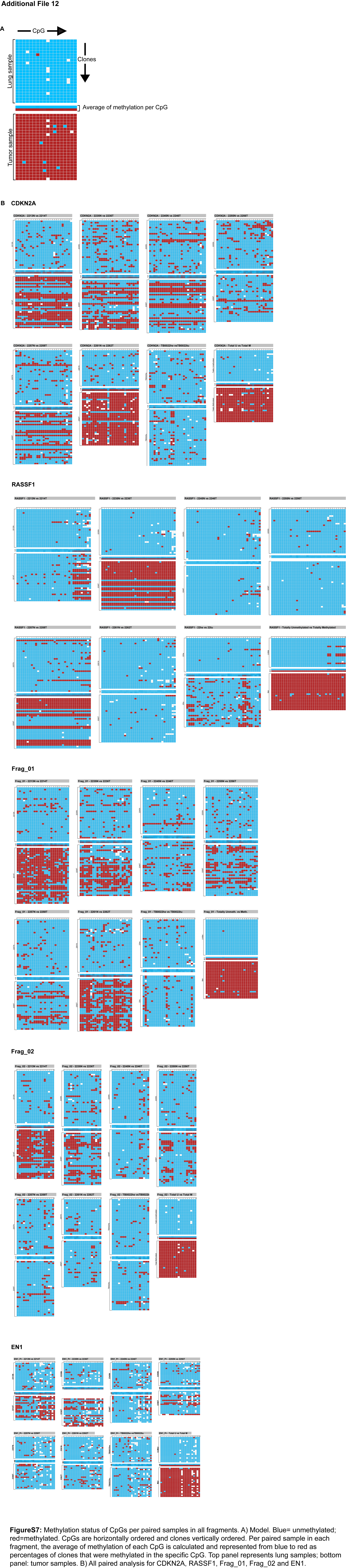
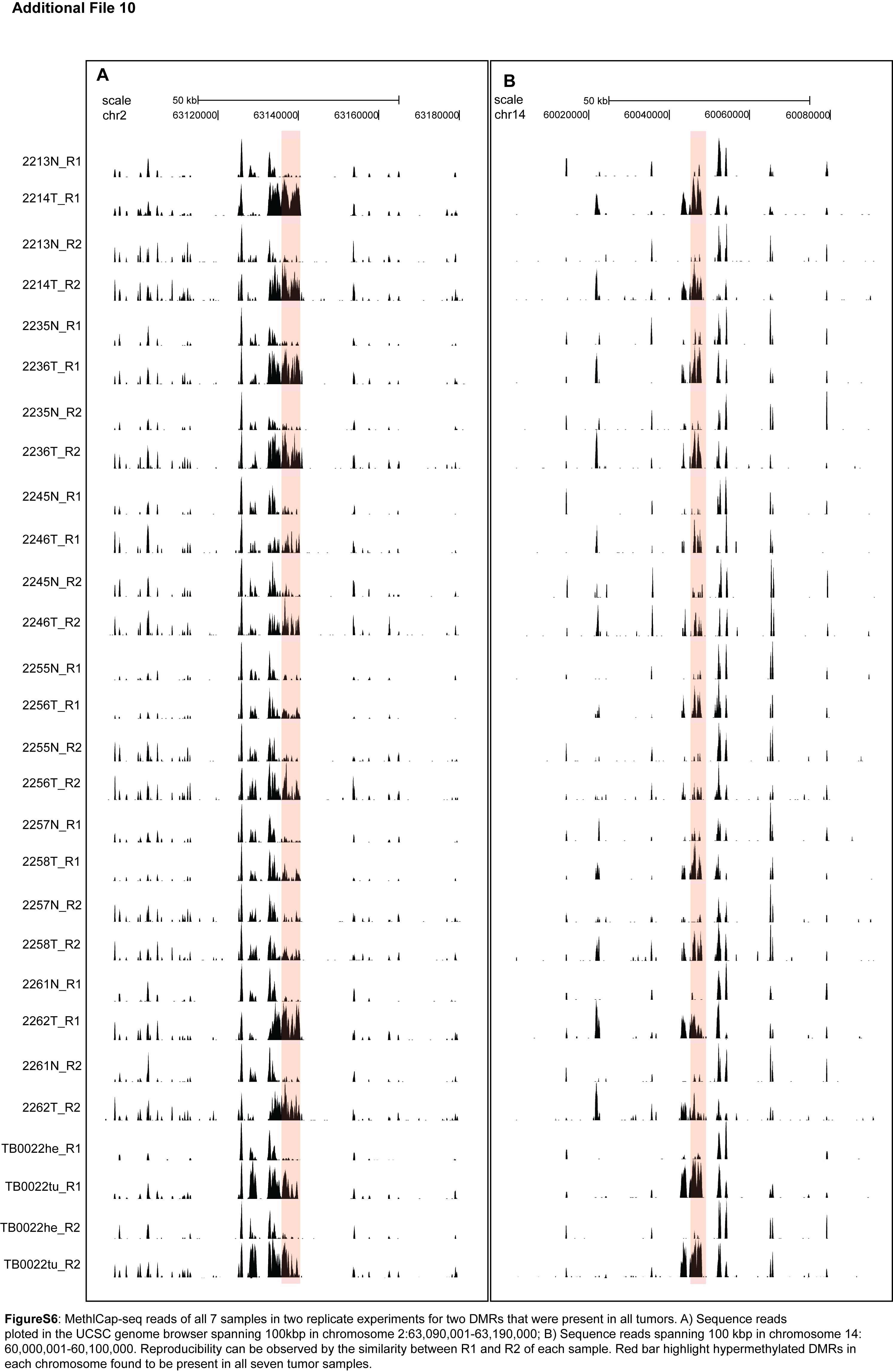
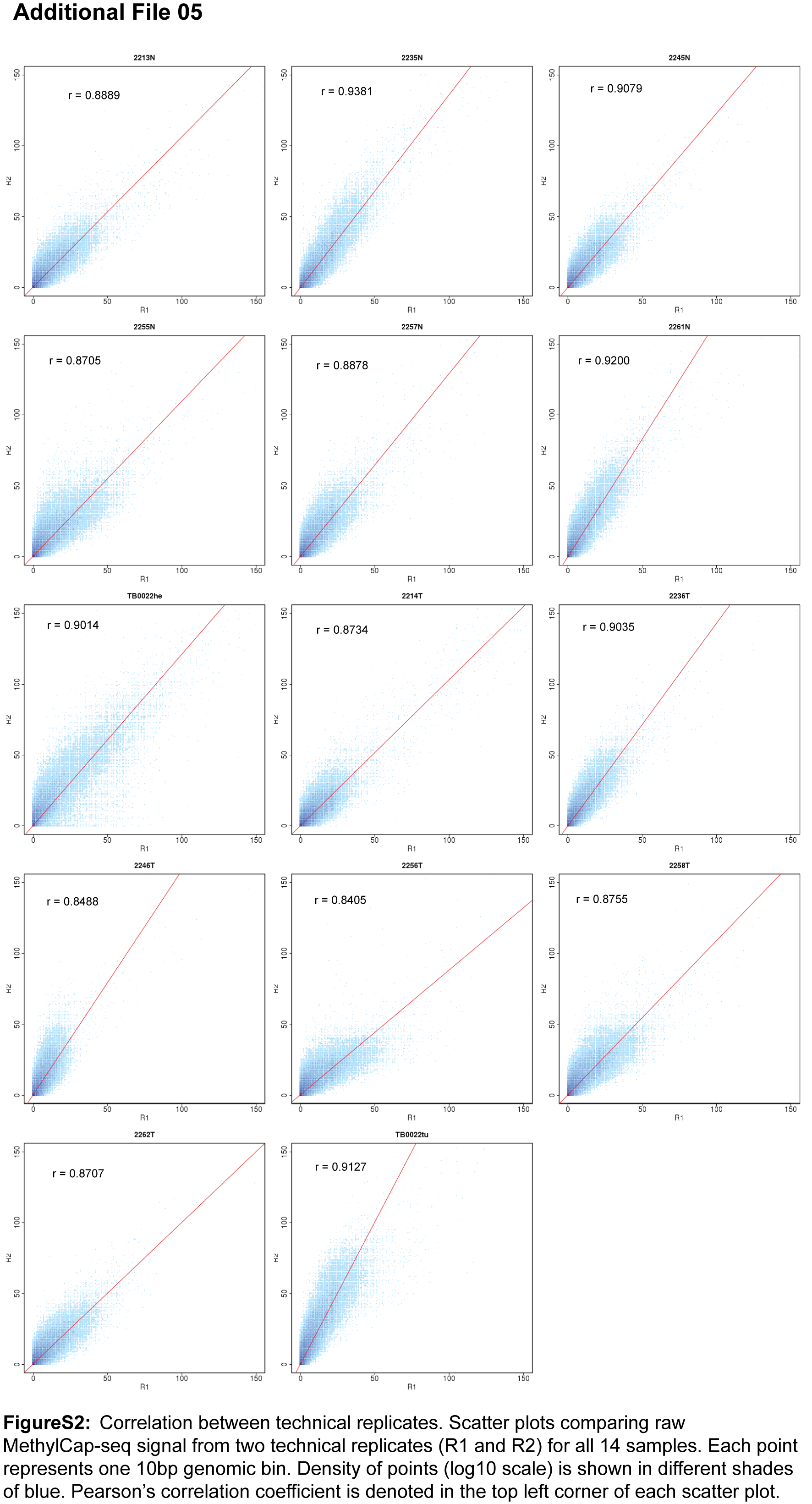
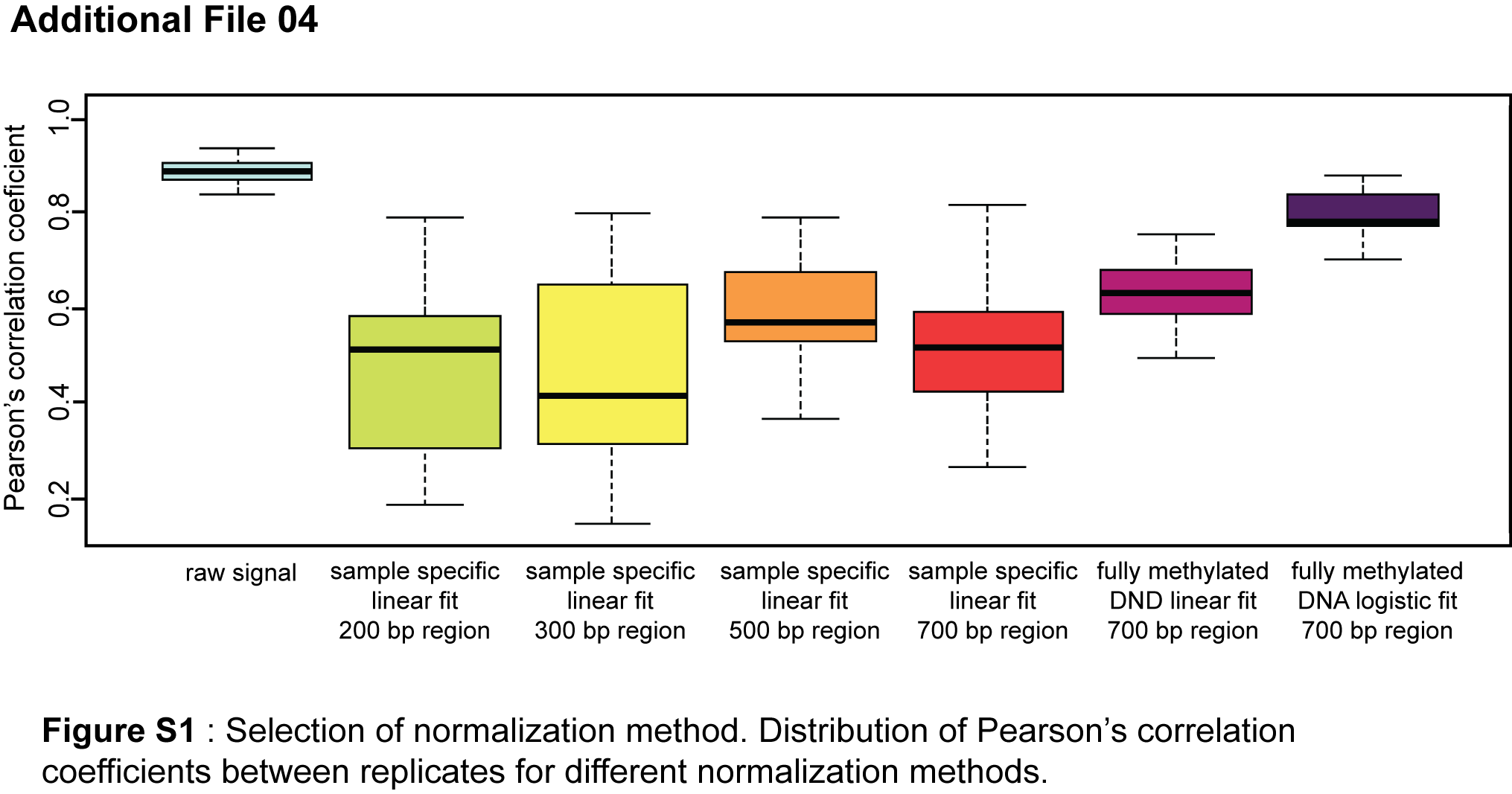
Background: Non-small cell lung carcinoma (NSCLC) is a complex malignancy that owing to its heterogeneity and poor prognosis poses many challenges to diagnosis, prognosis and patient treatment. DNA methylation is an important mechanism of epigenetic regulation involved in normal development and cancer. It is a very stable and specific modification and therefore in principle a very suitable marker for epigenetic phenotyping of tumors. Here we present a genome-wide DNA methylation analysis of NSCLC samples and paired lung tissues, where we combine MethylCap and next generation sequencing (MethylCap-seq) to provide comprehensive DNA methylation maps of the tumor and paired lung samples. The MethylCap-seq data were validated by bisulfite sequencing and methyl-specific polymerase chain reaction of selected regions. Results: Analysis of the MethylCap-seq data revealed a strong positive correlation between replicate experiments and between paired tumor/lung samples. We identified 57 differentially methylated regions (DMRs) present in all NSCLC tumors analyzed by MethylCap-seq. While hypomethylated DMRs did not correlate to any particular functional category of genes, the hypermethylated DMRs were strongly associated with genes encoding transcriptional regulators. Furthermore, subtelomeric regions and satellite repeats were hypomethylated in the NSCLC samples. We also identified DMRs that were specific to two of the major subtypes of NSCLC, adenocarcinomas and squamous cell carcinomas. Conclusions: Collectively, we provide a resource containing genome-wide DNA methylation maps of NSCLC and their paired lung tissues, and comprehensive lists of known and novel DMRs and associated genes in NSCLC.
Utgiver
BioMed CentralTidsskrift
Epigenetics & ChromatinOpphavsrett
Rejane Carvalho et al.; licensee BioMed Central Ltd.Copyright 2012 Hughes Carvalho et al.; licensee BioMed Central Ltd

Med mindre annet er angitt, så er denne innførselen lisensiert som Attribution CC BY